
- 产品描述
峦灵是行业内**的咨询公司,汇聚了来自法规、等各领域对法规有深刻理解的人士及*,致力为企业提供一站式、*、系统化的技术咨询服务, 帮助企业向**市场提供安全、有效和合规的产品。
欧洲体外诊断(IVD)CE标志
在欧洲销售的所有体外诊断(IVD)器械都需要CE标记。CE标记意味着IVD器械符合欧洲体外诊断器械指令(IVDD 98/79/EC),并且该器械可能在欧盟合法上市。欧洲新的体外诊断法规(IVDR)将于2022年强制实施,从而改变了IVD的监管要求。
技术文件或设计文档包括有关的设计、功能、组成、制造过程、使用、声称和估的详细信息。它们是所有类别的设备(I类,I类无菌,I类测量,IIa,IIb和III类)所必需的,但是由于器械的种类和涉及的制造、估过程的差异,没有两个文件是相同的。
CE技术文件或设计文档(Class III)是相当于国内(中国)在产品上市前递交给药监局进行审的注册文件,是对所涉及的一个综合全面的的描述,旨在表明产品符合欧洲指令的要求。因此,编写产品的技术文件或设计档案是欧洲CE认证过程中的非常关键的步骤,特别是在目前欧洲和公告机构对技术文档的监管和估日益加严的背景下,制造商准备的CE技术文件的质量往往成为CE认证的**和瓶颈。
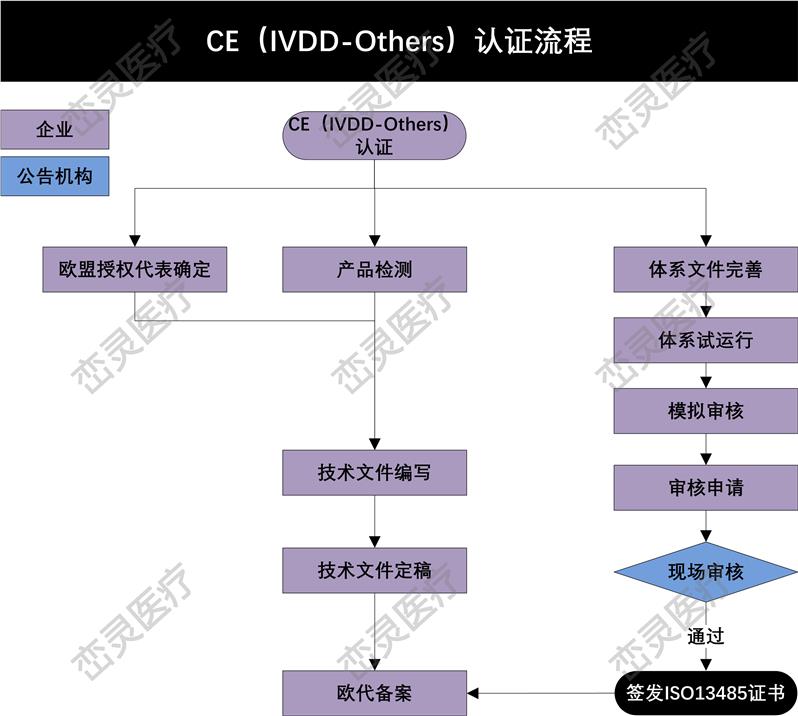
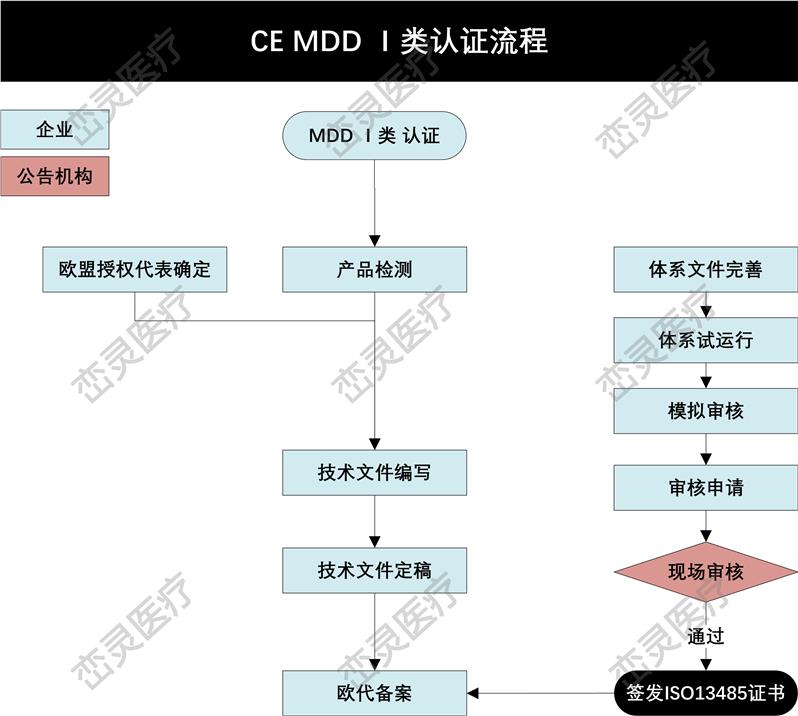
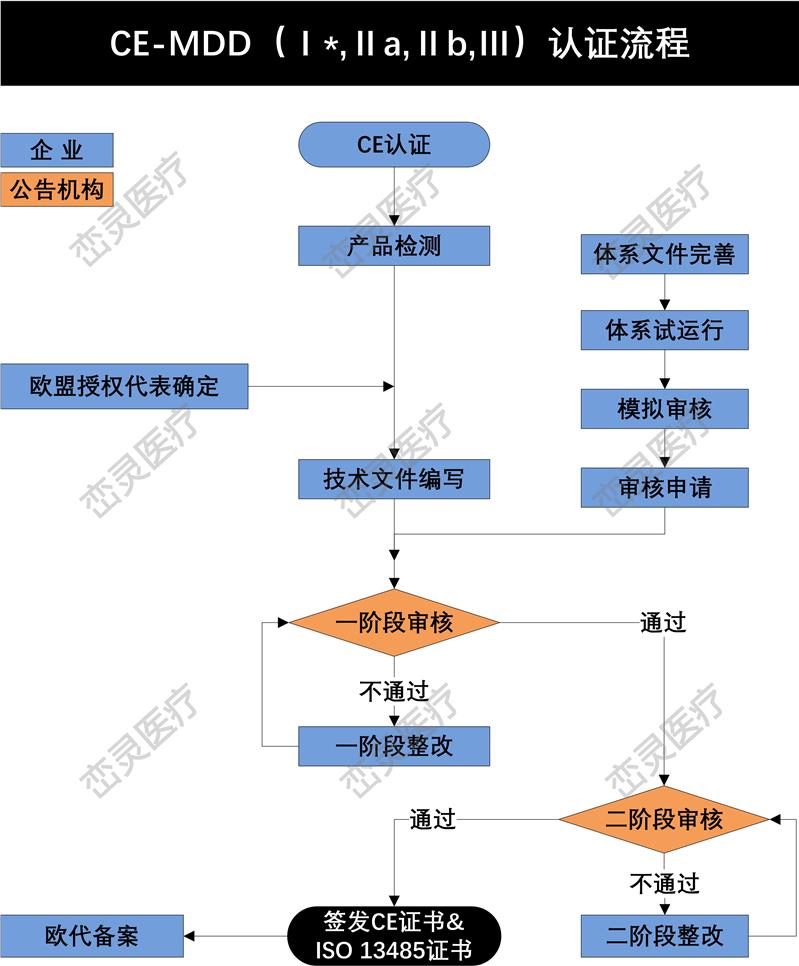
1. 确定根据指令的定义,产品是否属于,哪个欧盟指令适用于所考虑的:指令(93/42 / EEC),体外诊断器械指令(98/79/EC)或有源植入式指令(90/385 / EEC)
2. 确定的分类。
3. 实施质量管理体系(如适用)。大多数公司使用ISO 13485来满足要求。
4. 准备CE标记的技术文件(Technical Documentation)或设计文档(Design Dossier)。
5. 如果制造商在欧洲没有实际的场地,需要任命一位欧洲授权代表,代表制造商在欧盟境内进行相关的法规联络事宜。
6. 制造商的质量管理体系(QMS)和技术文件/设计文档由公告机构(Notified Body)审核和估。若器械属于I类非灭菌、I类无测量功能或顾客定制产品,则不需要公告机构的介入。
7. 从公告机构处获得CE证书和ISO 13485证书。
8. 起草符合性声明(DoC),其中所涉及的符合相应的指令。
9. 在产品上(包括标签、说明书等)加贴CE标志,并投放欧洲市场。对部分欧洲国家,可能需要在上市前在该国主管当局处进行备案。
10. 维持质量体系和CE证书的有效性。
我们的服务包括:
法规咨询认证:
欧盟 CE 认证(MDD/MDR/IVDD/IVDR , CE 欧盟授权代表以及产品注册和自由销售证书);
中国 NMPA 注册(Ⅰ器械备案,Ⅱ /Ⅲ类注册,认证,产品测试检验, 试验);
美国 FDA 注册(510K文档撰写与认证,产品列名、工厂注册、美国代理人、UDI的合规咨询与服务);
峦灵是行业内的咨询公司,汇聚了来自法规、等各领域对法规有深刻理解的人士及*,致力为企业提供一站式、*、系统化的技术咨询服务, 帮助企业向**市场提供安全、有效和合规的产品, 迄今为止,我们已与众多公司开展合作, 协助他们将安全、有效、合规的产品迅速推向**市场。 我们的服务包括: 法规合规服务(CE认证、NMPA注册、FDA注册、**注册) 质量管理体系(ISO13485、NMPA GMP、MDSAP、FDA QSR820、质量体系日常维护服务); 测试服务(有源产品测试认证咨询、无源产品测试认证咨询、清洗消毒灭菌验证确认、定制化摸底测试服务) **注册(加拿大、澳大利亚、日本、巴西、韩国、俄罗斯、东南亚注等); 法规培训服务(企业定制培训、小班课培训)
欢迎来到上海峦灵医疗信息技术有限公司网站,我公司位于历史文化悠久,近代城市文化底蕴深厚,历史古迹众多,有“东方巴黎”美称的上海市。 具体地址是上海青浦公司街道地址,负责人是阮贞。
主要经营CE认证 FDA510K。
你有什么需要?我们都可以帮你一一解决!我们公司主要的特色服务是:CE认证,FDA注册,**注册等,“诚信”是我们立足之本,“创新”是我们生存之源,“便捷”是我们努力的方向,用户的满意是我们最大的收益、用户的信赖是我们最大的成果。
本页链接:http://www.cg160.cn/vgy-52826467.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
关于上海峦灵医疗信息技术有限公司
商铺首页 |
更多产品 |
联系方式
峦灵是行业内的咨询公司,汇聚了来自法规、等各领域对法规有深刻理解的人士及*,致力为企业提供一站式、*、系统化的技术咨询服务, 帮助企业向**市场提供安全、有效和合规的产品, 迄今为止,我们已与众多公司开展合作, 协助他们将安全、有效、合规的产品迅速推向**市场。 我们的服务包括: 法规合规服务(CE认证、NMPA注..
- 我要给“柳州欧盟自由销售证书”留言
- 更多产品












相关分类






