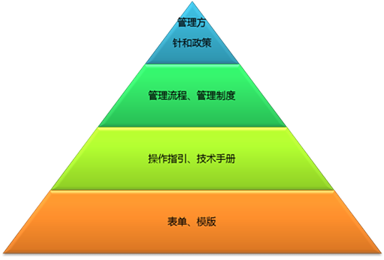
- 产品描述
-
深圳汉墨管理咨询有限公司是经广东省深圳市工商局批准并注册登记的具有法人资格的综合管理咨询公司。公司具有丰富的培训咨询人才资源并组建了各个管理模块的培训咨询团队,致力于通过培训或咨询帮助企业解决管理困惑,如:体系认证培训与咨询、社会责任验厂培训与咨询、质量管理培训与咨询、现场管理培训与咨询,精益生产项目辅导、企业管理诊断等。公司始终坚持“从实际出发,着眼于未来”的宗旨,为选择了我们公司的客户提供**来需求考虑的符合企业实际的培训咨询方案。

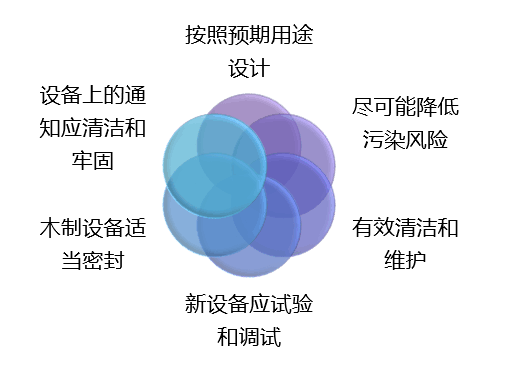
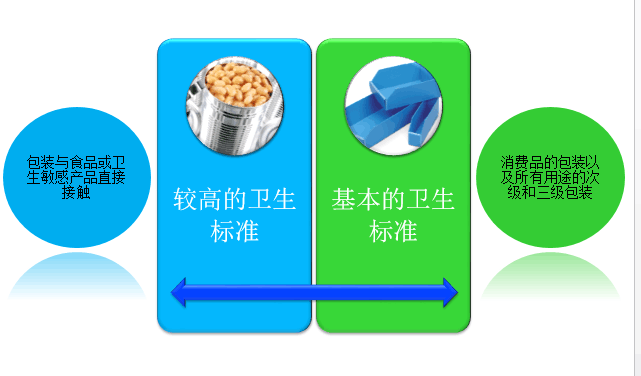
服务区域
服务广东省珠三角、粤东、粤西和粤北四个区域,其中珠三角:广州、深圳、佛山、东莞、中山、珠海、江门、肇庆、惠州;粤东:汕头、潮州、揭阳、汕尾;粤西:湛江、茂名、阳江;粤北:韶关、清远、云浮、梅州、河源。
汉墨提供以下认证及培训辅导咨询项目(包括但不限于):
1、认证与验厂(质量、食品安全、环境、安全等): ISO9001(ISO9000)质量管理体系认证、ISO22000食品安全管理体系认证、BRC食品安全**标准认证、IFS、HACCP危害分析与关键控制点管理体系认证、FSSC22000食品安全体系认证、ISO14001环境管理体系认证、ISO45001职业健康与安全管理体系认证、IATF16949汽车质量管理体系认证、ISO50001能源管理体系认证、ISO13485、ISO22716、GMP良好操作规范认证、FSC森林COC产销监管链体系认证、GAP良好农业规范认证、BSCI社会责任验厂、SEDEX、WRAP、SA8000、体系内审员培训等。
2、管理培训:管理自我(以客户为中心、情绪管理、执行力、个人行为特征、自我认知),管理业务,管理他人(MTP中高层管理能力提升、目标与计划管理、以经营为导向的**绩效、团队管理、冲突管理、辅导下属)等。
3、管理咨询:组织架构梳理及人才战略、**力测评与发展服务、培训管理体系搭建、企业文化梳理与落地等。
您有任何疑问,请随时与我们联系!
欢迎来到深圳汉墨管理咨询有限公司网站,我公司位于经济发达,交通发达,人口密集的中国经济中心城市—深圳。 具体地址是广东深圳龙岗区公司街道地址,负责人是林先生。
主要经营ISO9001认证。
你有什么需要?我们都可以帮你一一解决!我们公司主要的特色服务是:商务服务 认证服务 等,“诚信”是我们立足之本,“创新”是我们生存之源,“便捷”是我们努力的方向,用户的满意是我们最大的收益、用户的信赖是我们最大的成果。
本页链接:http://www.cg160.cn/vgy-101073057.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
关于深圳汉墨管理咨询有限公司
商铺首页 |
更多产品 |
联系方式
深圳汉墨管理咨询有限公司是经广东省深圳市工商局批准并注册登记的具有法人资格的综合管理咨询公司。公司具有丰富的培训咨询人才资源并组建了各个管理模块的培训咨询团队,致力于通过培训或咨询帮助企业解决管理困惑,如:体系认证培训与咨询、社会责任验厂培训与咨询、质量管理培训与咨询、现场管理培训与咨询,精益生产..
- 我要给“清远GMP认证顾问”留言
- 更多产品












相关分类






