
- 产品描述
一.医疗器械CE认证(MDD指令)概述
欧盟为消除各成员国间的贸易壁垒,逐步建立成为一个统一的大市场,以确保人员、服务、资金和产品(如医疗器械)的自由流通。在医疗器械领域,欧盟会制定了三个欧盟指令,以替代原来各成员的认可体系,使有关这类产品投放市场的规定协调一致。
这三个指令分别是:
1. 有源植入性医疗器械指令(AIMD, 90/335/EEC),适用于心脏起搏器,可植入的泵等有源植入性医疗器械。AIMD于1993年1月1日生效。过渡截止期为1994年12月31日,从1995年1月1日强制实施。
2. 活体外诊断器械指令(IVD),适用于血细胞计数器,妊娠检测装置等活体外诊断用医疗器械。该指令目前仍在起草阶段,可能于1998年末或1999年初正式实施。
3. 医疗器械指令(Medical Devices Direc-tive,93/42/EEC),适用范围很广,包括除有源植入性和体外诊断器械之外的几乎所有的医疗器械,如无源性医疗器械(敷料、一次性使用产品、接触镜、血袋、导管等);以及有源性医疗器械,如核磁共振仪、超声诊断和治疗仪、输液泵等。
二.医疗器械CE认证(MDD指令)**要求:
(一)基本要求(总要求)
a)安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析);
b)风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统);
c)性能符合性(产品的基本要求);
d)器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。);
e)器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响)。
(二)基本要求的具体包括如下14条:
1. 器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人们所接受,但应具有高水平的防护办法。
2. 生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应:
首先:应尽可能降低甚至避免危险
其次:对无法避免的危险采取适当的防护措施,包括安装报警装置;
*后,告知用户所提供防护措施的弱点及其可能带来的危险。
3. 器械必须**生产者期望获得的功能。器械设计制造和包装应有利于条(2)(A)D多规定的各项功能的发挥。
4. 在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3款所指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。
5. 器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会发生根本逆变。
6. 的大小同器械的使用性能相比较可以为人们所接受。
7. 化学、物理和生物性能
8. 感染和微生物污染。
9. 组装和环境因素
10. 检测器械
11. 辐射防护
12. 带有能源或与其他能源相连接的器械
13. 生产者提供的操作信息
14. 如果需要根据医疗数据确定器械是否满足基本要求,如*六款的情形,有关数据必须按照附录Ⅹ的规定**。
三.医疗器械CE认证(MDD指令)产品分类
医疗器械指令附录九中详定18条规则,按医疗产品的危险程度,将产品分为Ⅰ类、Ⅱa类、Ⅱb类、Ⅲ类。
产品分类规则:
1、规则应用由器械的预期使用目的决定;
2、如果器械是和其它器械配合使用,分类规则分别适用于每种器械;
3、附件可以和其它一起使用的器械分开单独分类;
4、启动或影响某种器械的软件与器械属于同一类型。
分类准则:
时 间: 暂时 (<60分钟)、短期(<30天)、长期(>30天)
创伤性:非创伤、通过孔径创伤,外科创伤 、植入。
适用位置:循环、**神经系统,其它地方。
能量供应:无源,有源。
规则1~4、所有非创伤性器械均属于I类,除非他们:
用于储存体液(血袋例外) II a类
于Ila类或较高类型的有源医疗器械类 II a类
改变体液成分 II a/II b类
一些伤口敷料 II a/II b类
规则5、侵入人体孔径的医疗器械
暂时使用(牙科压缩材料、检查手套) I类
短期使用(导管、隐形眼镜) II a类
长期使用(正常牙线) II b类
规则6-8、外科创伤性器械
再使用的外科器械(钳子,斧子) I类
暂时或短期使用(缝合针。外科手套) 11a类
长期使用(假关节,眼内晶体) II b类
与循环系统(CCS)或**神经系统接触的器械 III类
规则13、与医用物质结合的器械(含杀的避孕套、含抗生素的牙髓材料) III类
规则14、避孕用具(避孕套、帽 II b类 ) II b/III类 (内避孕器III类)
规则15、清洗或消毒的器械
医疗器械(内窥镜消毒) II a类
接触镜(消毒液、护理液) II a类
规则16、用于记录X射线图象的器械(X光片) II a类
规则17、利用动物组织的器械(生物)心脏瓣膜、肠线、胶原) III类
规则18、血袋 II b类
规则9、给予或交换能量的**器械 II a类
(肌肉器、电钻、皮肤光疗机、助听器)一一种潜在危险方式工作的 IIb类
(婴儿培养箱、高频电刀、超声碎石机、X光机)
规则10、诊断器械
提供能量(核磁共振,超声诊断仪) II a类
诊断/监视体内放射药物分布 II a类
(r照相机、正电子**成像仪)
诊断/监视生理功能(心电图、脑电图) II a类
危险情况下监视生理功能 II b类
(手术中的血气分析仪)
发出电离辐射(X射线诊断议) II b类
规则11 控制药物或其他物质进出人体的有源器械 II a类
(吸引设备、供给泵)
如以一种潜在危险方式工作 II b类
(麻醉机、呼吸机、透析机、高压氧舱)
规则12.所有其他有源医疗器械属于I类
(观察灯、牙科椅、轮椅、牙科用**灯、记录处理观察诊断图象用的有源器械)
四.医疗器械CE认证(MDD指令)技术文档(TCF)要求
“技术文档"是欧盟医疗器械指令中很重要的一个事项,它的目的是要求企业准备充份的技术资料和证明,供主管机关抽查,或发生诉讼纠纷时使用。医疗器械指令MDD 93/42/EEC要求"技术档案"可能包含下列项目:
A. 企业的质量手册和程序文件
B. 企业简介及欧洲授权代表名称、联系方式
C.CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的证明材料)
1. 产品名称、分类及引用标准条款的简要描述
2. 产品概述(包括类型和预期用途)
a) 产品的历史沿革
b) 技术性能参数
c) 产品配合使用的附件、配合件和其它设备清单
d) 产品的图示与样品
e) 产品所用原材料及供应商
3. 使用该产品的调和标准/或其它标准
4. 风险分析评估结论和预防措施(ISO14971产品服务危险分析报告)
5. 生产质量控制
a) 产品资料和控制文档(包括产品生产工艺流程图)
b) 产品的灭菌方法和确认的描述
c) 灭菌验证
d) 产品质量控制措施
e) 产品稳定性和效期的描述
6.包装和标识
a) 包装材料说明
b) 标签
c) 使用说明书
7. 技术评价
a) 产品检验报告及相关文献
b) 技术概要及*观点
8. 潜在风险评价
a) 产品潜在风险测试报告及相关文献
b) 潜在风险的概要及*观点
9. 临床评价
a) 产品临床测试报告及相关文献
b) 临床使用概述及*观点
附录1、产品出厂检测报告
附录2、产品型式检测报告
附录3、基本要求检查表
注:
1. 临床研究(包括:物理性能,生化、药理 、药动及毒性研究,功效测试,灭菌合格证明,药物相容性等)
2. 生物兼容性测试(A)EN30993 部分要求:细胞毒性、感光性、-皮内反应、急性全身中毒、致热性、亚急性中毒、遗传毒性、植入溶血性;B)支持测试:慢性 中毒、致性、再生性/生长性、生物动因退化。)
3. 临床资料(需要临床研究或描述临床研究)
4. 包装合格证明(EN868)
5. 标签、使用说明(EN980、EN1041)
6. 结论(设计档案资料的接受、利益对应风险的陈述)
七、技术文件的要求
MDR中添加了对技术文件内容的要求;且明确指出上市后监管计划和安全性较新报告(PSUR)都是技术文件的一部分,并要求依据上市后监管体系收集的资料对技术文件中相应信息进行较新。
八、技术文件的基本内容
器械说明与性能指标
包括变型和附件包含器械说明与性能指标,以及引用的前代和类似器械的信息。
制造商提供的信息
设计与制造信息
通用安全与性能要求
包含其符合附录I提供的通用安全与性能要求的证明资料。
风险利益分析和风险管理
产品验证与确认
临床前和临床数据(包含临床评价计划/报告,PMCF计划/报告);以及针对含药器械、人体/动物来源组织或其物制备的器械、引入人体并被吸收器械、具有测量功能器械等的相关附加信息
九、上市后监管的技术文件
AnnexIII TECHNICAL DOCUMENTATION ON POST-MARKET SURVEILLANCE 详细说明了要按照Article83-86 编写上市后监管的文件,包含上市后监管计划、上市后监管报告或定期安全性较新报告(PSUR)。
十、符合性声明文件
ANNEX IV EU DECLARATION OF CONFORMITY 详细说明了“符合性声明”文件包含的内容。
十一、加强器械上市后监管体系
Chapter VII POST-MARKET SURVEILLANCE, VIGILANCE AND MARKET SURVEILLANCE 着重说明上市后监管、警戒和市场监管。
建立、实施和维护上市后监管体系(见Article83)。
强调上市后监管体系贯穿整个生命周期,并不断较新。
建立“上市后监管计划”(见Article84),具体内容见Annex III。
I类器械编写“上市后监管报告”(见Article85)。
IIa、IIb和III类器械编制“定期安全性较新报告(PSUR)”(见Article86)。
PSUR需定期较新并作为技术文件的一部分。
建立警戒和上市后监管电子系统(见Article 92)。
在整个器械使用寿命期间,依据实施PMCF后**的临床数据对临床评价及技术文件进行较新(Annex XIV part B)。
十二、完善临床评价相关要求
新法规提出:
要求根据Article61和附录XIV partA执行、评估、报告和较新临床评价资料;
提出对特定III类和IIb类器械,CER中要考虑咨询*小组的意见;
对植入和III类器械,提出考虑临床研究;
要求CER按照PMCF**数据进行较新;
针对III类和可植入器械,提出了CER较新的频率;
明确证明实质等同性需考虑的特点;
要求其与风险管理的相互作用
十三、Eudamed数据库
新法规提出:
明确欧洲医疗器械数据库(Eudamed)建立目的和包含的信息(Article 33);
信息的公开性:
要求III类器械和植入式器械,安全和临床性能信息通过Eudamed向公众开放。
十四、提出器械的可追溯性(UDI)
除定制和研究器械外,其他器械均需建立UDI系统;
UDI信息体现在标签或包装上(不包含集装箱);
UDI-DI信息需要载明于符合性声明中(见Article27);
Annex VI Part B提出UDI-DI包含的信息;
可植入、重复使用、软件、可配置器械的UDI有特殊要求(见Annex VI Part C)
包装或标签上UDI实施的时间见Article123 (f)。
UDI 发行实体由欧盟会*。
过渡性:Article 120指出“在会根据*27(2)条*发行实体前,GS1、HIBCC和ICCBBA应被视为*的发行实施
办理ce认证需要准备哪些资料,费用大概多少
CE的含义:
CE”标志是一种安全认证标志,被视为制造商打开并进入欧洲市场的护照。凡是贴有“CE”标志的产品就可在欧盟各成员国内销售,无须符合每个成员国的要求,从而实现了商品在欧盟成员国范围内的自由流通。 在欧盟市场“CE”标志属强制性认证标志,不论是欧盟内部企业生产的产品,还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志,以表明产品符合欧盟《技术协调与标准化新方法》指令的基本要求。这是欧盟法律对产品提出的一种强制性要求。
CE标志的意义在于:
用CE缩略词为符号表示加贴CE标志的产品符合有关欧洲指令规定的主要要求(Essential Requirements),并用以证实该产品已通过了相应的合格评定程序和/或制造商的合格声明,真正成为产品被允许进入欧共体市场销售的通行证。有关指令要求加贴CE标志的工业产品,没有CE标志的,不得上市销售,已加贴CE标志进入市场的产品,发现不符合安全要求的,要责令从市场收回,持续违反指令有关CE标志规定的,将被限制或禁止进入欧盟市场或退出市场。
近日欧盟了一份针对于一类医疗器械CE MDR申请步骤的MDCG文件(MDCG 2019-15),今天我就和大家来解读一下该文件的要求。文件中总共提到了八个步骤,今天我们先讲步骤0-3。
第零步指的就是厂商要做的准备。这一步的主要工作是要把MDR的要求整合到现有的质量管理体系中。怎么理解呢?在做CE MDR认证之前厂商已经建立了质量管理体系,现在要做CE认证,要在原来的质量管理体系的基础上增加MDR的特殊要求,要形成相应的文件(包括程序文件和三级文件)。
举个例子,MDR的条款87给出了严重不良事件上报的要求,那么厂商就要基于该条款的要求形成相应的程序文件。
步是确认你的产品到底是不是医疗器械,这个非常简单。主要去看条款2定义中的个定义,个定义就是医疗器械的定义,厂商需要去判断一下你的产品符不符合这个定义。一般的产品是很好判断的,但是也有一些边界(Borderline)产品,你可能需要基于一些指南文件去判断。这份MDCG的指南文件提到欧盟的主管机构需要这些指南文件。
第二步是确认你的产品的分类,看它是不是一类。这个工作主要去参考MDR的附录VIII,需要注意的是原来在MDD中算是I类医疗器械的产品现在由于分类规则的变化(比如新增了独立软件产品的规则11,新增了涉及到进入人体的产品的规则22),在MDR中的分类也会发生变化(可能就不是I类了)。
步和第二步相对来说是容易的,接下来我们来看看工作颇多的第三步,第三步涉及到的主要工作包括:
a)确认是否满足MDR附录I的基本安全和性能要求
附录I的基本安全和性能要求分三个章节,总共有23个条款。比老法规MDD的附录I多了9个条款。章节的基本要求增加了风险管理系统和使用错误的要求。*二章设计和生产的要求增加了药械组合产品,人源和动物源材料产品,医疗软件(包括独立软件和嵌入式软件),有源植入器械,非专业人士使用器械风险的要求;标签和说明书的要求被放到了*三章,要求内容更多了较细了。
b)进行临床评价
临床评价的部分目前和MDR配套的指南文件还没有,I类医疗器械厂商可以按照MEDDEV 2.7/1 Rev 4.0的要求去准备。需要提醒一点的是,可能有一些I类器械的临床评价工作是不需要提供临床数据的(比如,检查手套),这种情况MDR的法规也是允许的,但是厂商要提供合理的说明(MDR条款61(10))。
c)准备技术文件
厂商要按照MDR附录II和附录III的要求准备技术文件。技术文件的要求也是MDR和MDD差异较大的比方,MDD中没有一个附录的要求是专门针对技术文件的,但MDR有俩。
d)和公告机构提出申请(对于I类灭菌,I类测量和I类可重复使用的器械)
I类产品只有三种情况才需要公告机构介入发证,分别是I类灭菌,I类测量和I类可重复使用。其它情况厂商只要愉快地做自我声明就可以了。MDR条款52(7)有提到厂商可以选择以下符合性评价途径:附录IX的章和*三章、附录XI的A部分。同样在条款52(7)提到,公告机构的审核应局限于以下三点:
对于I类灭菌产品,关注如何建立,保证和维持产品的无菌保证水平。
对于I类测量产品,关注产品如何满足计量的要求,比如采用的单位是否符合要求,测量的准确性和精密度如何保证。
对于I类可重复使用的医疗器械,关注清洗,消毒,灭菌,维护和功能测试,还有产品的说明书。
e)准备产品标签和说明书
我建议厂商可以按照MDR附录I的*三章去准备标签和说明书,适用的内容都要有,不适用的地方要说明理由。
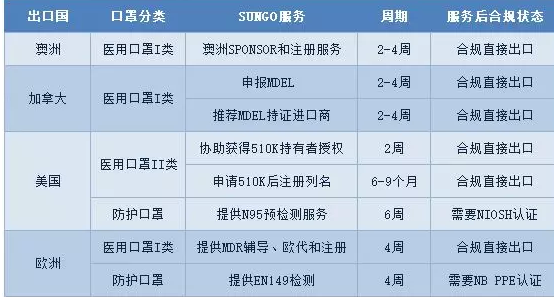
SUNGO集团是**化的医疗器械法规专业技术服务商。 SUNGO品牌包括六家公司,分别是位于美国的SUNGO Technical Service Inc.,位于英国的SUNGO Certification Company Limited,位于荷兰的SUNGO Europe B.V.,位于澳洲的SUNGO Australia,位于德国的SUNGO Cert GmbH和上海沙格企业管理咨询有限公司(含上海总部、武汉分公司和广州分公司)。
欢迎来到上海沙格企业管理咨询有限公司网站,我公司位于世界上面积最大的红松原始林所在地—伊春市。 具体地址是黑龙江伊春金山屯公司街道地址,负责人是袁阳。
主要经营FDA注册和FDA510K。
你有什么需要?我们都可以帮你一一解决!我们公司主要的特色服务是:商务服务 认证服务 等,“诚信”是我们立足之本,“创新”是我们生存之源,“便捷”是我们努力的方向,用户的满意是我们最大的收益、用户的信赖是我们最大的成果。
本页链接:http://www.cg160.cn/vgy-54372319.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
关于上海沙格企业管理咨询有限公司
商铺首页 |
更多产品 |
联系方式
SUNGO集团是**化的医疗器械法规专业技术服务商。 SUNGO品牌包括六家公司,分别是位于美国的SUNGO Technical Service Inc.,位于英国的SUNGO Certification Company Limited,位于荷兰的SUNGO Europe B.V.,位于澳洲的SUNGO Australia,位于德国的SUNGO Cert GmbH和上海沙格企业管理咨询有限公司(含上海总部、武汉分公司和..
- 我要给“清远口罩CE认证提供什么资料”留言
- 更多产品








相关分类






