
- 产品描述
峦灵欧盟市场准入整体解决方案
服务内容包括CE技术文档撰写、辅导、测试、认证方案。
还包括欧代服务、欧盟FSC、WEEE、Rohs、ISO14971 风险分析、评价、灭菌、软件周期、可用性等欧盟合规的咨询与服务。
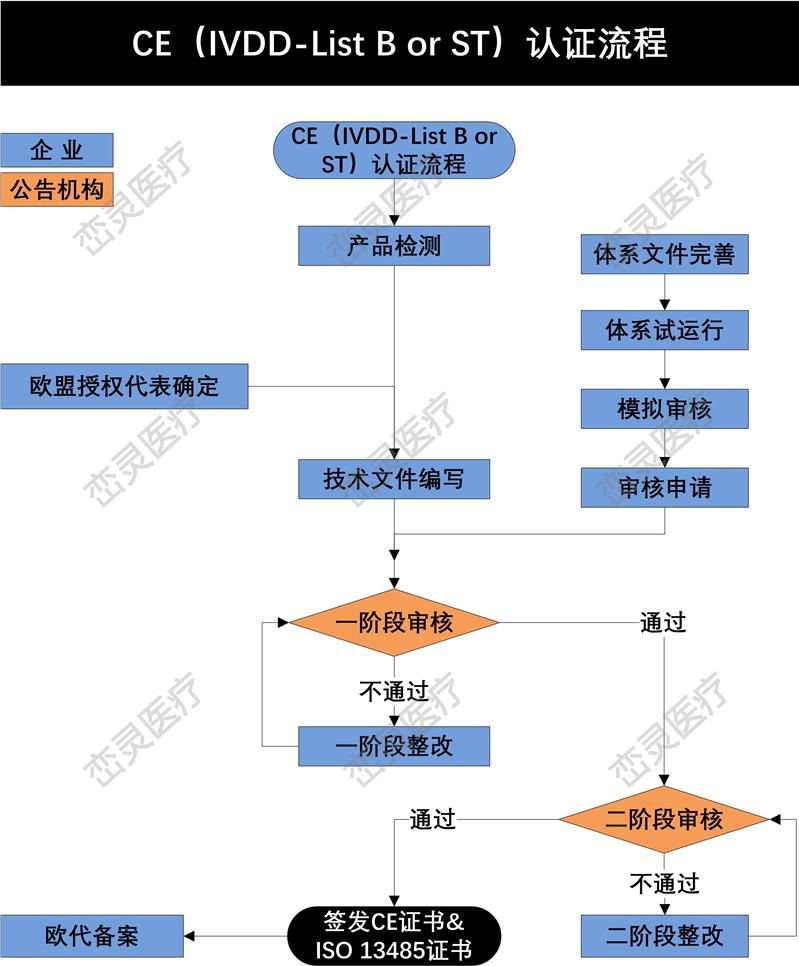
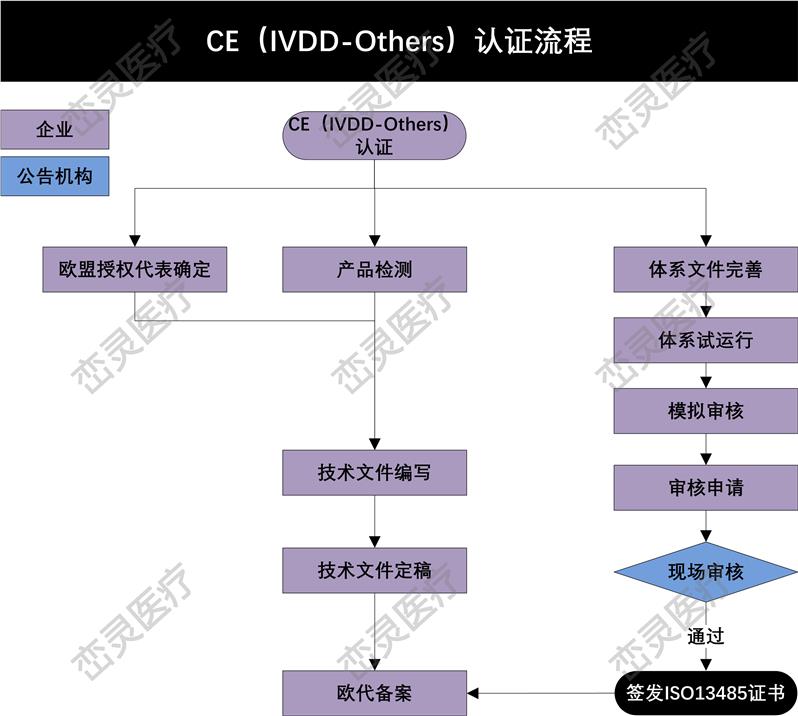
MDD及MDR对技术文件的要求 – 技术文件编制的方式
有一个基本的概要来描述各主要内容, 并索引至所有相关文件及存放地
只有概要文件存放在同一地点
被索引的相关文件需存放在索引*的地点
文件的版本能在*的地点找到
定义每一个地点的归档系统
MDR附录III 上市后监督技术文件
MDR中上市后监督的定义(Article2(60)): “上市后监管”是指制造商与其他经济运营商合作开展的所有活动 ,目的旨在建立并保持的系统化程序,以主动收集和总结从已投放市场、市场上可获得或投入使用的器械获得的经验,以确定是否需要立即采取任何必要的纠正或预防措施。
MDR符合性声明
MDR: 独立附录IV
- 制造商、欧盟授权代表的名称和地址;
- 声明制造商对符合性声明担负责任;
- 基本的UDI-DI信息;
- 器械分类
- 器械的名称、规格型号;
- 所符合的法规和采用的符合性评定路径;
- 进行符合性评定的公告机构的名称、地址和机构代码;
- 声称符合的通用规范(CS);
- 符合性声明的签署日期和地点;
- 具有法律效力的签名以及被授权人的职能。
对于III类产品的技术文件(MDD时的设计文档)要求和其他类别的没有差异。制造商需要提供一套十分完整的技术文件。
我们的服务包括:
器械法规咨询认证:
欧盟 CE 认证(MDD/MDR/IVDD/IVDR , CE 欧盟授权代表以及产品注册和自由销售证书);
中国 NMPA 注册(Ⅰ器械备案,Ⅱ /Ⅲ类器械注册,认证,产品测试检验, 试验);
美国 FDA 注册(510K文档撰写与认证,产品列名、工厂注册、美国代理人、UDI的合规咨询与服务);
其他国家的认证注册咨询:包括**法规注册咨询服务,如澳大利亚、新西兰、加拿大、巴西、俄罗斯、日本、韩国等**国家的注册咨询服务。
器械测试咨询服务:
有源产品测试、无源产品测试、定制化摸底测试、测试整改;
器械质量管理体系咨询服务:
NMPA GMP、ISO 13485、FDA QSR820、MDSAP体系培训、建立及运行辅导。

峦灵是行业内的咨询公司,汇聚了来自法规、等各领域对法规有深刻理解的人士及*,致力为企业提供一站式、*、系统化的技术咨询服务, 帮助企业向**市场提供安全、有效和合规的产品, 迄今为止,我们已与众多公司开展合作, 协助他们将安全、有效、合规的产品迅速推向**市场。 我们的服务包括: 法规合规服务(CE认证、NMPA注册、FDA注册、**注册) 质量管理体系(ISO13485、NMPA GMP、MDSAP、FDA QSR820、质量体系日常维护服务); 测试服务(有源产品测试认证咨询、无源产品测试认证咨询、清洗消毒灭菌验证确认、定制化摸底测试服务) **注册(加拿大、澳大利亚、日本、巴西、韩国、俄罗斯、东南亚注等); 法规培训服务(企业定制培训、小班课培训)
欢迎来到上海峦灵医疗信息技术有限公司网站,我公司位于历史文化悠久,近代城市文化底蕴深厚,历史古迹众多,有“东方巴黎”美称的上海市。 具体地址是上海青浦公司街道地址,负责人是阮贞。
主要经营CE认证 FDA510K。
你有什么需要?我们都可以帮你一一解决!我们公司主要的特色服务是:CE认证,FDA注册,**注册等,“诚信”是我们立足之本,“创新”是我们生存之源,“便捷”是我们努力的方向,用户的满意是我们最大的收益、用户的信赖是我们最大的成果。
本页链接:http://www.cg160.cn/vgy-52854847.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
关于上海峦灵医疗信息技术有限公司
商铺首页 |
更多产品 |
联系方式
峦灵是行业内的咨询公司,汇聚了来自法规、等各领域对法规有深刻理解的人士及*,致力为企业提供一站式、*、系统化的技术咨询服务, 帮助企业向**市场提供安全、有效和合规的产品, 迄今为止,我们已与众多公司开展合作, 协助他们将安全、有效、合规的产品迅速推向**市场。 我们的服务包括: 法规合规服务(CE认证、NMPA注..
- 我要给“常州MDR公告机构”留言
- 更多产品












相关分类






