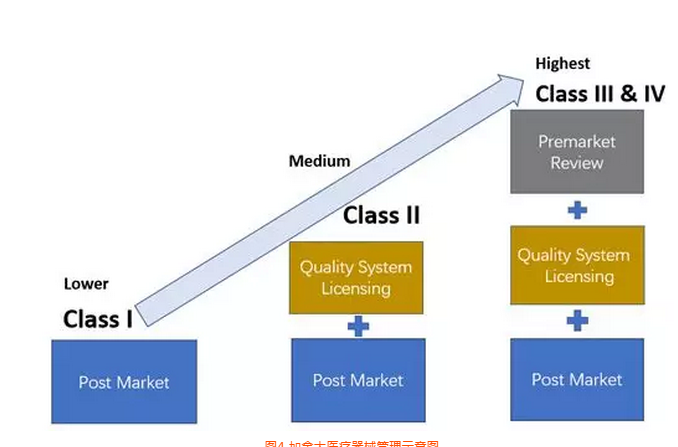
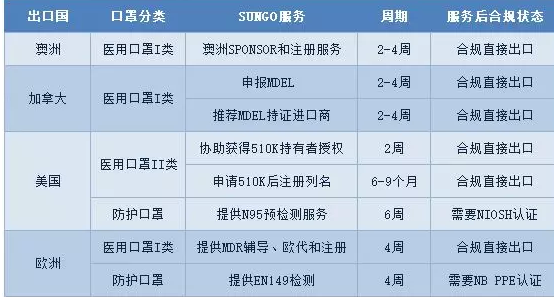
- 产品描述


SUNGO集团是**化的医疗器械法规专业技术服务商。 SUNGO品牌包括六家公司,分别是位于美国的SUNGO Technical Service Inc.,位于英国的SUNGO Certification Company Limited,位于荷兰的SUNGO Europe B.V.,位于澳洲的SUNGO Australia,位于德国的SUNGO Cert GmbH和上海沙格企业管理咨询有限公司(含上海总部、武汉分公司和广州分公司)。
欢迎来到上海沙格企业管理咨询有限公司网站,我公司位于世界上面积最大的红松原始林所在地—伊春市。 具体地址是黑龙江伊春金山屯公司街道地址,负责人是袁阳。
主要经营FDA注册和FDA510K。
你有什么需要?我们都可以帮你一一解决!我们公司主要的特色服务是:商务服务 认证服务 等,“诚信”是我们立足之本,“创新”是我们生存之源,“便捷”是我们努力的方向,用户的满意是我们最大的收益、用户的信赖是我们最大的成果。
本页链接:http://www.cg160.cn/vgy-51664034.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
关于上海沙格企业管理咨询有限公司
商铺首页 |
更多产品 |
联系方式
SUNGO集团是**化的医疗器械法规专业技术服务商。 SUNGO品牌包括六家公司,分别是位于美国的SUNGO Technical Service Inc.,位于英国的SUNGO Certification Company Limited,位于荷兰的SUNGO Europe B.V.,位于澳洲的SUNGO Australia,位于德国的SUNGO Cert GmbH和上海沙格企业管理咨询有限公司(含上海总部、武汉分公司和..
- 我要给“海口医用口罩CE认证提供什么资料 速度快”留言
- 更多产品









相关分类






