- 产品描述
CE认证中REGULATION (EU) 2017/745,简称“MDR”
根据TUV莱茵新消息,5月份以后将不再接MDD指令的CE订单,且现在接的订单企业必须在8月31日之前接受审核。
由此可见,接下来将会是II类CE的一个申请高峰期,毕竟MDR的费用会比MDD高很多。
2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。依据MDR Article 123的要求,MDR于2017年5月26日正式生效,并与2020年5月26日期正式取代MDD(93/42/EEC)和AIMDD(90/385/EEC)。
CE技术文件是欧盟器械指令中很重要的一个事项,它的目的是要求企业准备充份的技术资料和,供主管机关抽查,或发生诉讼纠纷时使用。各欧盟指令对于"技术档案"的要求有所差别,在这里谨以中国出口企业常用的“器械”的要求为例,加以说明。
器械MDR法规要求"技术档案"可能包含:企业的质量手册和程序文件;企业简介及欧洲代表名称、联系方式;CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的材料),主要内容如下:
1、产品名称、分类
2、产品概述(包括类型和预期用途)
◇产品的历史沿革
◇技术性能参数
◇产品配合使用的附件、配合件和其它设备清单
◇产品的图示与样品
◇产品所用原材料及供应商
3、使用该产品的调和标准/或其它标准
4、风险分析评估结论和预防措施
5、生产质量控制
◇产品资料和控制文档(包括产品生产工艺流程图)
◇产品的灭菌方法和确认的描述
◇灭菌验证
◇产品质量控制措施
◇产品稳定性和效期的描述
6、包装和标识
◇包装材料说明
◇标签
◇使用说明书
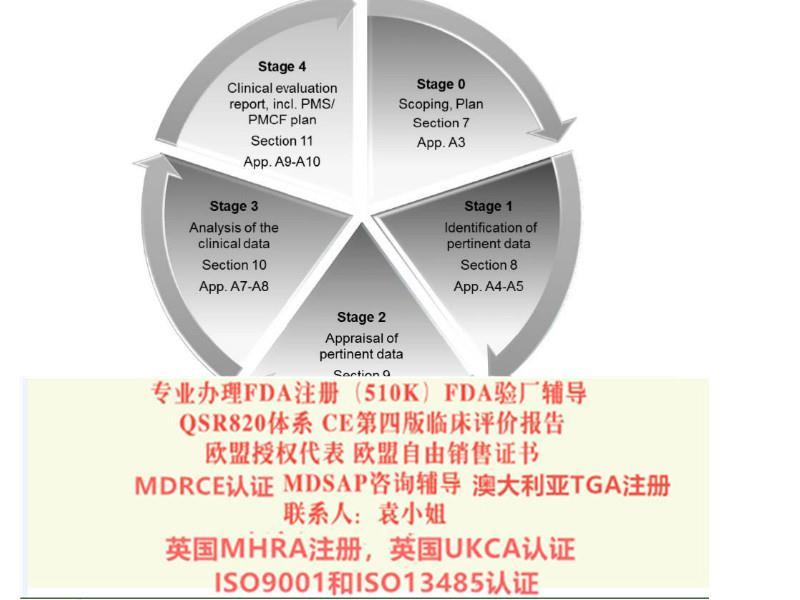
7、技术评价
◇产品检验报告及相关文献
◇技术概要及观点
8、风险管理
◇产品潜在风险报告及相关文献
◇潜在风险的概要及观点
9、评价
◇产品测试报告及相关文献
◇使用概述及观点
附1、产品出厂检测报告
附2、产品型式检测报告
附3、基本要求检查表
2017年5月5日欧盟就发布了新版器械法规MDR(EU 2017/745)。在2017年5月25日,MDR正式生效。老的器械指令即MDD( 93/42/EEC)与新的MDR(EU 2017/745)指令的交替过渡期为三年。
也就是说从2020年5月26日, MDR指令在欧盟就将开始强制执行,它将完全取代过去老的器械指令MDD (93/42/EEC)和老的有源植入器械指令AIMDD(90/385/EEC)。
需要特别指出的是:
☑ MDR强制执行后,新申请的CE认证必须按照MDR执行;
☑ 当前没有CE证书的产品,自5月27日起,必须按照MDR认证;
☑ 2020年5月26前签发的MDD证书,在有效期内仍然可以用,晚到2024年5月26日;
☑ 原有MDD证书需在证书失效前换发 MDR。
1. 本次爆发期间所获CE认证的医用口罩,可以说95%以上的都是按照老的MDD指令进行的,可能要面临新版换证问题;
2. 根据目前欧盟统计,拥有老指令版本MDD(93/42/EEC)授权的NB公告机构共有56家,而符合MDR授权的NB公告机构目前则仅有12家而已。也就是说,从2020年5月26日开始,针对医用口罩的CE认证审核机构可选性降低了80%;
3. 由于欧盟MDR此类授权审核机构(NB:Notified Body)可选性的减少,必然造成医用口罩CE认证费用相当大的概率将有大幅提升的可能;
4. 新版MDR指令审核要求比老版MDD指令更为复杂,认证周期必然大幅度拉长,本次期间有些机构声称的几天出证的可能性将基本为零;
5. 获得CE认证的口罩等设备必须要求有的欧盟授权代表(简称"欧代"),这在以前MDD指令时对一些低风险产品其实有没有欧代并不严格,但在MDR指令后,即使是在一些电商平台上进行销售的产品也会要求提供必要的欧代信息;
6. MDR的体系审核流程和要求更为复杂与繁琐,举个例子如在MDR条款15中要求,器械厂商应在其组织架构内,至少配备一名负责合规的人员,即合规负责人(Person responsible for regulatory compliance)。该人员应具备器械领域的必要知识,并且有一系列的性要求(如法律、、、工程或其他相关学科,并且至少拥有一年与器械法规事务或质量管理体系相关的经验)。
7. 对于已经在欧盟渠道正式上市的产品来说,老的MDD指令CE证书可以保持到2024年5月26日。但如果企业产品在今年5月26日前,并未在欧盟市场销售的,原则上在今年5月26日后应该将老的MDD证书重新申请调整到MDR版本。
欧洲议会和理事会于2017年4月5日颁布了关于器械的新法规(EU)2017/745,并将在2020年5月26日取代之前的MDD指令和IVDD指令。 ----Regulation (EU) 2017/745 on Medical Devices
2020年4月23 日,欧盟理事会和议会对器械MDR法规进行了修订。规定将MDR法规的实施日期推迟一年,至2021年5月26日。
MDR较之MDD变化主要表现在:
强化制造商的责任:合规负责人/持续更新技术文件/财务保障。
更严格的上市前评审:部分产品的分类变高/加强对证据的要求/对特定高风险器械将采用上市前审查机制,由欧盟级别的组参与,进行更严格的事先评估。
适用范围扩大:非用途,但其功能和风险特征与器械相似的器械将同样纳入MDR 的管理范围。
提高透明度和可追溯性:使用器械标识(UDI)系统识别和械/患者将收到具有所有基本信息的植入卡/将建 立包含器械认证信息和研究、警戒和上市后监测信息的修订后的可公开访问的EUDAMED 数据库。
SUNGO公司介绍 SUNGO GROUP: SUNGO TECHNICAL SERVICE INC美国公司; SUNGO Certification Company Limited英国公司 SUNGO Europe B.V.荷兰公司; SUNGO Cert GmbH德国公司; SUNGO Australia澳大利亚公司; 上海沙格企业管理咨询有限公司() 上海沙格企业管理咨询有限公司武汉分公司 上海沙格企业管理咨询有限公司广东分公司 SUNGO集团凭借全球网络和专业队伍为全球客户提供法规,在医疗器械行业尤为专长。 我们可以为您提供的自主服务项目主要有: 出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表、欧盟自由销售证书、防护服PPE指令Type5/6认证 出口美国法规:医疗器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂辅导及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系辅导、食品FDA验厂辅导及整改、OTC品FDA验厂辅导及整改 法规:医疗器械产品备案登记表、医疗器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证辅导、SFDA验厂辅导、SFDA注册检测、企业标准编制、监局自由销售证。 出口其余国际法规:医疗器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂辅导、ISO22716 GMPC验厂辅导、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试 上海沙格企业管理咨询公司已经在国内近千家客户提供了相关服务,也受美国几家大型采购集团委托,对其在的供应商进行二方审核工作,我们的客户集中在无纺布行业企业、敷料耗材、病床、轮椅、体外诊断医疗器械行业、大型饲料机械设备企业、电子电器行业类别。公司一直秉承“专业辅导、增值服务、、本土价格”的方针,为客户提供优质的服务。 我们服务过的部分知名企业及上市公司有: 新华医疗、鱼跃医疗、威高集团、阳普医疗、艾迪尔医疗、驼人医疗、恒安集团、康德莱集团、阿蓓纳、上海联影、上海复旦医疗、微创骨科医疗、普罗医学、凯利泰、联医医疗、羚锐制、江西3L、正昌集团、杭州可靠、台钜集团、维达集团、青岛光电、科勒、暴龙眼镜等等
欢迎来到上海沙格企业管理咨询有限公司网站,我公司位于历史文化悠久,近代城市文化底蕴深厚,历史古迹众多,有“东方巴黎”美称的上海市。 具体地址是上海浦东陆家嘴公司街道地址,负责人是袁玲。
主要经营ISO13485认证。
本公司以科技为前导,质量为中心,全员为基础,依国际水准,创智博品牌。质量、信誉是公司生存和发展的基石;ISO9000质量体系为保证。选择我们的产品:咨询 管理咨询 ,是正确的决定!
本页链接:http://www.cg160.cn/vgy-109405477.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
- 产品推荐
- 金华气体腐蚀试验CNAS/CMA认证 CNAS/CMA认证 绍兴做N95认证注册申请哪家公司好-口罩认证申请 内江塑料成分检测 提供快速48小时服务 湿巾杀灭病毒 电话多少 南通做N95认证注册申请价格-口罩认证申请 绍兴N95认证注册申请周期-口罩认证申请 连云港第三方检测机构-CNASCMA可靠性测试 第三方测评 南京第三方检测 检测机构 宿迁电子产品检测中心 第三方机构 戊二醛消毒剂检测怎么做 手消毒现场模拟 经验丰富 郑州TGA注册的资料 常州第三方检测机构可靠性测试 第三方检测机构
- 相关文章
- 医用帽2017/745 2017/745 / EU的CE认证升级指南纸尿裤2017/745 MDR医疗器械分类规则瑞代 吸唾管瑞代要求 MDRCE辅导要求腕式血压计的英代认证要求 什么是英国授权代表医用绷带英国代表 什么情况下需要提供英代床边扶手fda认证510k 美国FDA510K认证是什么 如何申请杨克连接管UKREP认证怎么申请 英国授权代表是什么棉签美国FDA510K认证 美国FDA510K认证是什么 如何申请电动救护车担架2017/745 医疗器械MDR法规纱布块MDRCE证书怎么申请 mdrce认证 评估包含什么注射器的UKREP认证怎么申请 英国授权代表是什么口罩MDSAP认证 怎么做
关于上海沙格企业管理咨询有限公司
商铺首页 |
更多产品 |
联系方式
SUNGO公司介绍 SUNGO GROUP: SUNGO TECHNICAL SERVICE INC美国公司; SUNGO Certification Company Limited英国公司 SUNGO Europe B.V.荷兰公司; SUNGO Cert GmbH德国公司; SUNGO Australia澳大利亚公司; 上海沙格企业管理咨询有限公司() 上海沙格企业管理咨询有限公司武汉分公司 上海沙格企业管理咨询有限公司广东分..
- 我要给“电动救护车担架CE MDR认证 详细流程步骤”留言
- 更多产品