- 产品描述
级医用防护服、隔离衣、手术衣,欧盟授权代表,签署《欧代协议》;
谁必须要申请FDA510(k)
FD&C Act的*510(k)规章中并没有特别指出谁必须申请510(k)——任何人都可以申请。但是,他们了哪种行为,例如把器械引入美国市场,要求510(k)申请。基于的行为,必须向FDA递交510(k)的有:
(1) 把器械引入美国市场的生产商;
(2) 把器械引入美国市场的研发设计者;
(3) 改变器械或器械标签的再包装者;
(4) 把器械引入美国市场的外国厂家/出口商或外国厂家/出口商的美国代理方
经济运营者是指制造商,授权代表,进口商,分销商以及系统或手术袋的任何组合或。投放市场的自然人或法人。也是说,负责按照法规生产设备(包括组合或),销售和上市后操作的自然人或法人2017 年4 月5 日,欧洲议会和理事会正式签发了欧盟关于器械*2017/745 号法规(MDR,EU2017/745),5月5日,欧盟(Official Journal of the EuropeanUnion) 正式发布该法规。
2017年5月5日欧盟发布OfficialJournal在这里我司需要特别说明的是欧盟此次是直接发布的Regulation(法规)而相比较之前的Directive(指令)其区别在于:提高了文件的约束力,发布立即在欧盟成员国生效并成为有约束力的法律,此次的Regulation*向Directive那样需要经过成员国转化成当地法律法规去落实实施。器械法规(MDR)转换期为3年,2020年5月4日起强制实行。
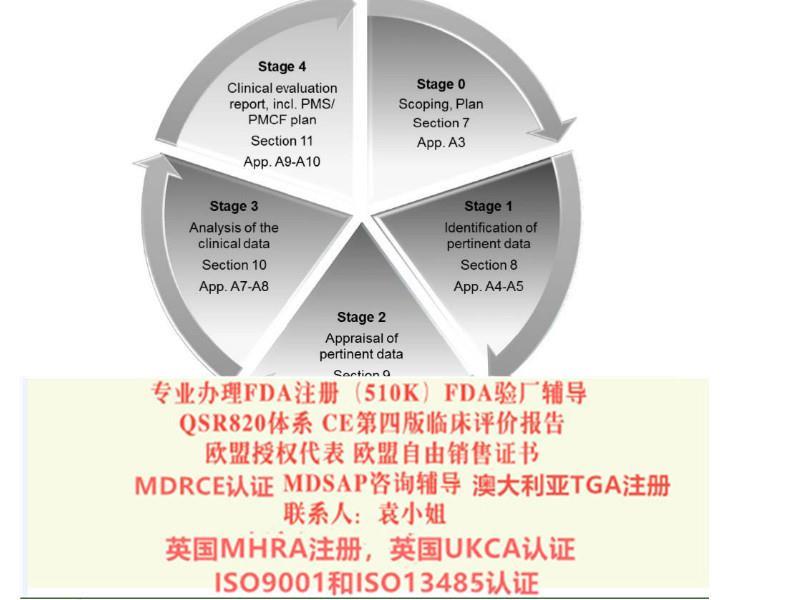
合格评定根据某些装置的危险类别和具体特征,对符合CE标志的装置的评价各不相同(*五十二条)。所有IIa、IIb和III类设备以及一些特定的I类设备都需要一个通知机构的干预(见*7a5、b6和c7段)。*52条和附件九、十、习叙述了根据设备类别不同的评估方法。在某些情况下,制造商对合格评定路线有一些选择。对于某些III类和IIb类设备,有一个新的评估咨询程序,该程序将由一个立的组根据该公告机构的评估评估报告进行(*五十四条)。附件一规定了一般的和性能要求,附件二和三规定了技术文件的组成。质量管理体系的范围(*10条*9款)包括评价和上市后随访。评价计划必须先于评价本身(附件十四,A部分)。
我公司办理: 出口英国需要UKCA认证,英国代表,MHRA注册



SUNGO品牌创建于2006年,立志于成为化的法规技术服务商。我司可以办理:1:欧盟MDR CE咨询,MDR欧盟授权代表,欧盟注册2:欧盟IVDR CE咨询,IVDR欧盟授权代表,欧盟注册3:美国FDA注册,FDA510K4:国内的注册证和生产证5:加拿大的MDEL注册6:ISO13485咨询和认证目前SUNGO在中国、欧洲、北美和澳洲均设有服务机构,服务过的客户较是覆盖了(中国、越南、马来西亚、孟加拉、新加坡)、欧洲(英国、瑞士、瑞典、丹麦、挪威)、北美(美国、加拿大)、南美(阿根廷)、大洋洲(澳大利亚)和非洲(博茨瓦纳、南非)等国家和地区。SUNGO致力于为的生产商和经营者提供市场准入的合规咨询以及**注册服务。从产品生产、检测、过程管理、注册、认证、整改、上市跟踪等各环节为企业提供的技术支持,为产品合规和顺利上市保驾**。十多年里,SUNGO已为30多家上市公司和制造商,合计5000多家企业提供过相关服务。SUNGO始终追求支持、服务和客户满意。所有客户都有一对一的客服对接以保持经常性的联系,提供在线即时服务,针对贸易中存在的技术壁垒方面的问题提供的支持和解。选择SUNGO,不是选择了一次性的合作伙伴,而是选择了一个长期的技术支持的伙伴。我们将秉承一贯“服务、客户”的原则,依托的技术团队,优化我们的服务,让更多的器械合法、安全进入市场,为器械行业健康发展贡献力量。因为,所以放心!
欢迎来到上海沙格企业管理咨询有限公司网站,我公司位于历史文化悠久,近代城市文化底蕴深厚,历史古迹众多,有“东方巴黎”美称的上海市。 具体地址是上海金山公司街道地址,负责人是袁小姐。
主要经营上海沙格企业管理有限公司SUNGO是欧盟授权代表,从事FDA验厂、欧盟自由销售证书、医疗器械单一体系审核MDSAP、CE*四版临床评价报告、MDRCE认证、CE MDR认证、CE技术文件、EU 201。
我们公司主要提供商务服务 认证服务 等服务,我们确信,凭借我们的专业服务和良好的协调、沟通能力,定能使客户在经营生产中无后顾之忧,协助客户不断成长,在合作中与客户实现共赢。欢迎您致电咨询!
本页链接:http://www.cg160.cn/vgy-105840486.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
- 产品推荐
- 青岛崂山区app开发公司定制-电话咨询 获取报价 肇庆墙体广告楼体喷绘美丽乡村绘画墙体挂布广告 南充体育场照明 照明检测 万宁公司注册型号 注册公司费用省 注册地址优惠 河源墙体广告喷绘写真广告制作形象墙彩绘高墙喷绘广告 茂名墙体广告喷绘广告墙美丽乡村壁画发布墙体广告 东莞黄江免抵退税哪里有 财税记账服务 做好账找九牛 整机 资阳金属油漆涂料机构 金相检测 出具报告 德阳室内亮度供应 教室灯光检测 韶关墙体广告承接墙体喷绘广告手绘墙画高墙喷绘广告 广东墙体广告墙体广告喷绘集装箱彩绘墙体刷墙广告 儋州公司注册 注册公司办理快 工商异常快速办
- 相关文章
- 胰岛素注射针清洗验证方案 UKCA证书 需要什么资料隔离衣的清洗验证方案 EC Rep 需要什么资料创可贴MHRA认证 MHRA注册 的关键纸尿裤MHRA认证 医疗器械产品MHRA注册 新规要求介绍颊面管MHRA注册 英国mhra注册 mhra如何申请电动救护车担架MHRA注册 MHRA认证注册 新规要求介绍手术衣的清洗验证方案 MDR CE证书 需要什么资料眼镜自由贸易证书 欧盟自由销售证书CFS 申请材料介绍病床自由销售证书 自由贸易证书 申请条件丁腈手套自由销售证书 CFS证书 有效期多久护具CFS证书 什么是欧盟自由销售证书 申请条件尿液分析仪美国FDA注册 什么是FDA注册流程
关于上海沙格企业管理咨询有限公司
商铺首页 |
更多产品 |
联系方式
SUNGO品牌创建于2006年,立志于成为化的法规技术服务商。我司可以办理:1:欧盟MDR CE咨询,MDR欧盟授权代表,欧盟注册2:欧盟IVDR CE咨询,IVDR欧盟授权代表,欧盟注册3:美国FDA注册,FDA510K4:国内的注册证和生产证5:加拿大的MDEL注册6:ISO13485咨询和认证目前SUNGO在中国、欧洲、北美和澳洲均设有服务机构,服务过..
- 我要给“温州袖套的MDRCE认证”留言
- 更多产品












相关分类






